Teil 2 – Datenimport, Datenbereinigung und Datenmanagement
06 · Datenbereinigung und Datentransformation
- Das Tidy-Data-Prinzip verstehen und erkennen, warum es Analysen vereinfacht.
- Fehlende Werte sehen, verstehen und begründet behandeln.
- Mit den fünf Kern-Verben filtern, auswählen, ableiten, gruppieren, zusammenfassen arbeiten.
- Zwei Tabellen über einen Join verbinden und zwischen long/wide umformen.
- Dasselbe Ergebnis in
pandas(Python) unddplyr/tidyr(R) erzeugen.
Auf dieser Seite
Klinischer Aufhänger
Du hast eine Aufnahmekohorte und separat die Laborwerte. Bevor du eine Frage beantworten kannst – „Haben septische Patient:innen ein höheres Laktat?“ – müssen die Daten sauber und zusammengeführt sein. Genau dieser Schritt kostet in der Praxis 80 % der Zeit. Wir machen ihn hier transparent.
1 Konzept: Tidy Data
Daten sind „tidy”, wenn
- jede Variable eine Spalte ist,
- jede Beobachtung eine Zeile ist,
- jede Beobachtungseinheit eine Tabelle bildet.
kohorte.csv ist bereits tidy (eine Zeile je Patient:in). vitalwerte.csv ist
tidy im long format (eine Zeile je Messung). Tidy Data ist die Grundlage, auf
der alle folgenden Module aufbauen, sauberer Input, verlässlicher Output.
Viele klinische Exporte kommen im wide-Format (eine Spalte je Messzeitpunkt). Frage dich vor jeder Berechnung: Ist das wirklich eine Variable pro Spalte? Eine Spalte „Laktat_Tag1” ist keine Variable, sie vermischt Merkmal und Zeitpunkt.
Für FortgeschritteneVertiefung
Im long-Format berechnet vitalwerte.csv Verläufe über alle
Tage mit einer einzigen groupby-Zeile, im wide-Format bräuchte es eine
manuelle Schleife über alle Tag-Spalten.
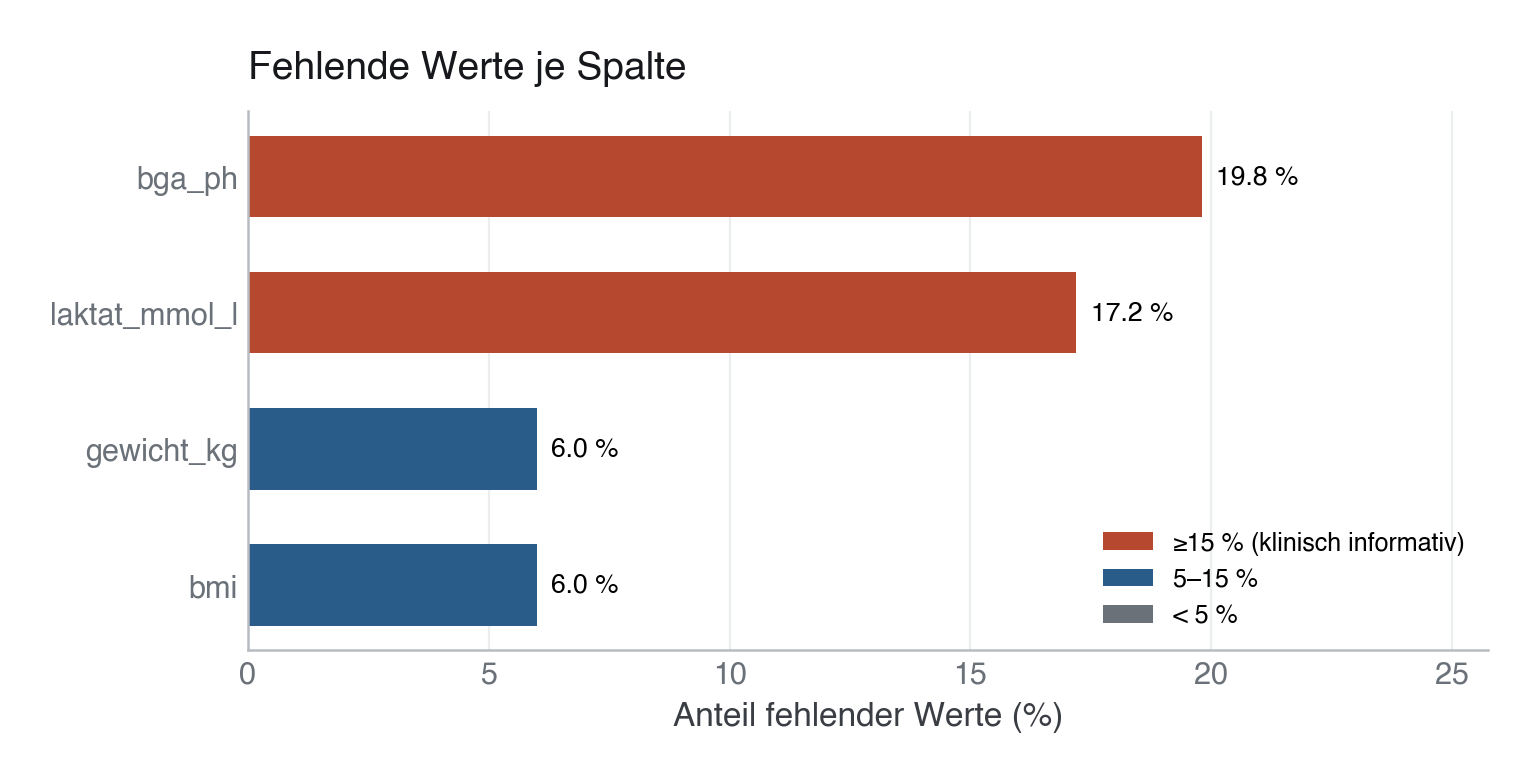
2 Fehlende Werte zuerst ansehen
Fehlende Werte nie „wegmachen”, bevor du sie verstanden hast. Erst zählen, dann entscheiden (löschen vs. behalten vs. imputieren). In unseren Daten fehlen drei Größen, aus drei verschiedenen Gründen:
| Spalte | fehlend | warum |
|---|---|---|
bmi, gewicht_kg |
~6 % | rein zufällig (MCAR) — kostet nur Power |
laktat_mmol_l |
~17 % | hängt vom sofa_score ab: Schwerkranke bekommen häufiger eine BGA |
bga_ph |
~20 % | hängt vom Outcome ab: bei früh Verstorbenen fehlt die Kontroll-BGA |
Die letzten beiden sehen gleich aus („nicht zufällig“) und haben völlig
verschiedene Konsequenzen. Beim Laktat hängt das Fehlen von einer beobachteten
Kovariate ab, nicht vom unbeobachteten Laktatwert selbst. Beim pH hängt es am
Outcome — und genau das entscheidet, ob dropna() unschädlich oder ein
Kunstfehler ist. Modul 14 rechnet beides vor.
dropna() ohne Argument löscht jede Zeile mit irgendeinem
fehlenden Wert. Auf unserem zusammengeführten Datensatz sind das 192 von 500
Patient:innen (38,4 %) — obwohl keine einzelne Spalte mehr als 20 % vermisst.
Die Verluste addieren sich über die Spalten. Gib stets explizit subset= an
oder prüfe vorher den Anteil fehlender Werte.
Für FortgeschritteneVertiefung
Das Muster der fehlenden Werte ist selbst eine klinische
Information. Weil die Laktat-Messwahrscheinlichkeit vom beobachteten
SOFA-Score abhängt, ist das Missing At Random (MAR), nicht MCAR (rein
zufällig wie bei bmi) und auch nicht MNAR (das würde bedeuten, die
Messwahrscheinlichkeit hinge vom Laktatwert selbst ab, den wir ja gerade nicht
kennen, wenn er fehlt). Beim bga_ph ist es ebenfalls MAR — aber abhängig vom
Outcome statt von einer Kovariate.
Das Etikett „MAR“ sagt für sich genommen nicht, ob eine Complete-Case-Analyse verzerrt. Entscheidend ist, wovon das Fehlen abhängt und ob diese Variable in deinem Modell steht. Modul 14 rechnet vor, dass Complete Case beim Laktat korrekte Koeffizienten liefert, beim pH aber ein um 28 % zu niedriges absolutes Risiko.
3 Die fünf Kern-Verben
| Aufgabe | pandas | dplyr |
|---|---|---|
| Zeilen filtern | df[df.x > 5] / .query() |
filter() |
| Spalten wählen | df[["a","b"]] |
select() |
| Spalte ableiten | df.assign(...) |
mutate() |
| gruppieren + zusammenfassen | groupby().agg() |
group_by() |> summarise() |
| Tabellen verbinden | merge() |
*_join() |
Der vollständige, lauffähige Code steht in code/python.py und
code/r.R. Hier die Kernidee:
In Python:
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.import pandas as pd cohort = pd.read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv") labs = pd.read_csv("https://schradern.github.io/data-science-coach/data/labor.csv") # 1) Standardise gender encoding ('w' -> 'weiblich') cohort["geschlecht"] = cohort["geschlecht"].replace({"w": "weiblich"}) # 2) Count missing values — understand before acting print(cohort.isna().sum()) # 3) Join: cohort + labs via patient_id df = cohort.merge(labs, on="patient_id", how="left") # 4) Group: median lactate by admission type print( df.groupby("aufnahmegrund")["laktat_mmol_l"] .median() .sort_values(ascending=False) )
…und dasselbe Ergebnis in R:
library(tidyverse) cohort <- read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv", show_col_types = FALSE) |> mutate(geschlecht = if_else(geschlecht == "w", "weiblich", geschlecht)) labs <- read_csv("https://schradern.github.io/data-science-coach/data/labor.csv", show_col_types = FALSE) df <- left_join(cohort, labs, by = "patient_id") df |> group_by(aufnahmegrund) |> summarise(laktat_median = median(laktat_mmol_l, na.rm = TRUE)) |> arrange(desc(laktat_median))
groupby().agg() ignoriert NaN standardmäßig, meist
gewünscht, aber bei kleinen Gruppen kann es still zu falschen n führen.
Gib value_counts() oder .size() parallel mit aus, um die tatsächliche
Gruppengröße zu kennen.
Für FortgeschritteneVertiefung
df.query("sofa_score >= 6 and aufnahmegrund == 'Sepsis'") ist
lesbarer als verkettete boolesche Masken, besonders bei vier oder mehr Bedingungen.
Zudem lässt sich query() direkt in eine Methodenkette integrieren.
4 Long ↔ Wide
vitalwerte.csv liegt long vor (eine Zeile je Messung). Manche Auswertungen
brauchen wide (eine Spalte je Tag). In pandas: pivot_table; in tidyr:
pivot_wider. Der umgekehrte Weg (melt / pivot_longer) ist genauso wichtig, die meisten Funktionen für Grafik und Statistik erwarten long.

.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.import pandas as pd vitals = pd.read_csv("https://schradern.github.io/data-science-coach/data/vitalwerte.csv") # Long -> Wide: one column per measurement day map_wide = vitals.pivot_table( index="patient_id", columns="tag", values="map_mmhg" ) print(map_wide.shape) # rows = patients, cols = days # Wide -> Long again (e.g. for plotting) map_long = map_wide.reset_index().melt( id_vars="patient_id", var_name="tag", value_name="map_mmhg" )
library(dplyr) library(tidyr) library(readr) vitals <- read_csv("https://schradern.github.io/data-science-coach/data/vitalwerte.csv", show_col_types = FALSE) # Long -> Wide map_wide <- vitals |> select(patient_id, tag, map_mmhg) |> pivot_wider(names_from = tag, values_from = map_mmhg, names_prefix = "tag") # Wide -> Long map_long <- map_wide |> pivot_longer(-patient_id, names_to = "tag", values_to = "map_mmhg")
pivot_table mit aggfunc="mean" (Standard) aggregiert
stillschweigend, wenn mehrere Zeilen pro Kombination existieren. Prüfen Sie,
ob tatsächlich nur eine Messung pro Patient:in und Tag vorliegt, sonst wählen Sie
aggfunc explizit und wissen, was zusammengefasst wird.
Für FortgeschritteneVertiefung
Bei mehreren Messvariablen (MAP, Herzfrequenz, Temperatur) lässt
sich pivot_wider(names_from = variable, values_from = wert) direkt auf ein
vollständiges long-Format anwenden, das ersetzt drei separate pivot-Aufrufe.
Fallstricke und Merksätze
- Erst zählen, dann handeln. Fehlende Werte sind eine Information, kein Müll.
na.rm = TRUE(R) bzw. pandas-skipnaändern Ergebnisse still, bewusst setzen.-
Ein
left_join/merge(how="left")darf die Zeilenzahl der linken Tabelle nicht erhöhen. Immer vorher/nachher die Form prüfen (df.shape). -
Tidy zuerst. Jede Stunde Aufräumen spart drei Stunden Analyse.
Selbstcheck
df.shape vorher und nachher prüfen.