Teil 5 – Machine Learning und KI in der Medizin
29 · Unüberwachtes Lernen und Phänotypisierung von Patient:innen
- Klinische Daten ohne Labels vorverarbeiten (Imputation, Standardisierung) und verstehen, warum Skalierung hier unverzichtbar ist.
- k-Means anwenden und k über Elbow-Kurve und Silhouettenkoeffizienten begründet wählen.
- Hierarchisches Clustering (Ward-Linkage) interpretieren und einen Dendrogramm lesen.
- PCA zur Dimensionsreduktion nutzen (erklärte Varianz, 2D-Projektion) und UMAP als nichtlineare Alternative einsetzen.
- Cluster zu klinischen Phänotypen profilieren und ehrlich kommunizieren, was sie nicht sind.
Auf dieser Seite
Klinischer Aufhänger
Nicht jede Forschungsfrage hat ein vordefiniertes Label. Manchmal lautet sie: Gibt es in unserer Kohorte distinkte Patientengruppen, die wir noch nicht kennen? Phänotypisierung, das datengetriebene Erkennen von Subgruppen, ist in Intensivmedizin, Sepsis-Forschung und Onkologie ein aktives Feld. Das Versprechen: unbekannte klinische Profile sichtbar machen. Die Gefahr: Artefakte für Biologie zu halten.
1 Vorverarbeitung ohne Zielvariable
Ohne Outcome-Label entscheiden allein die Merkmale über die Cluster. Fehlende Werte und unterschiedliche Skalen verzerren jeden Distanzalgorithmus.
Hinweis — die Codeblöcke unten sind Auszüge. Sie zeigen die entscheidenden Schritte, nicht jede Import-Zeile. Das vollständige, am Stück lauffähige Skript ist
code/python.py(im Browser über den Python-Reiter oben). In Colab führst du das ganze Modul mit einer Zeile aus:!python module/29-unueberwacht-phenotyping/code/python.py— siehe das in Colab öffnen.
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.import pandas as pd import numpy as np from sklearn.impute import SimpleImputer from sklearn.preprocessing import StandardScaler from sklearn.pipeline import Pipeline labs = pd.read_csv("https://schradern.github.io/data-science-coach/data/labor.csv") NUMERIC = ["alter", "sofa_score", "crp_mg_l", "bmi", "leukozyten_g_l", "kreatinin_mg_dl", "laktat_mmol_l"] df = pd.read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv").merge(labs, on="patient_id", how="left") X_raw = df[NUMERIC].values prep = Pipeline([ ("impute", SimpleImputer(strategy="median")), ("scale", StandardScaler()), ]) X = prep.fit_transform(X_raw)
Imputation und Skalierung erfolgen hier auf allen Daten, das ist korrekt, weil es keine Testmenge gibt, die wir schützen müssten. Trotzdem: Wer später einen neue:n Patient:in einem Cluster zuordnen will, muss denselben prep-Transformer wiederverwenden, nicht neu anpassen.
Für FortgeschritteneVertiefung
Bei kategorialen Merkmalen (z. B. aufnahmegrund) lieber Gower-Distanz oder One-Hot-Encoding mit nachgelagerter PCA verwenden, bevor k-Means läuft, euklidische Distanz auf Dummy-Variablen ist verzerrt.
Skalierungsabhängigkeit bei PCA und Clustering
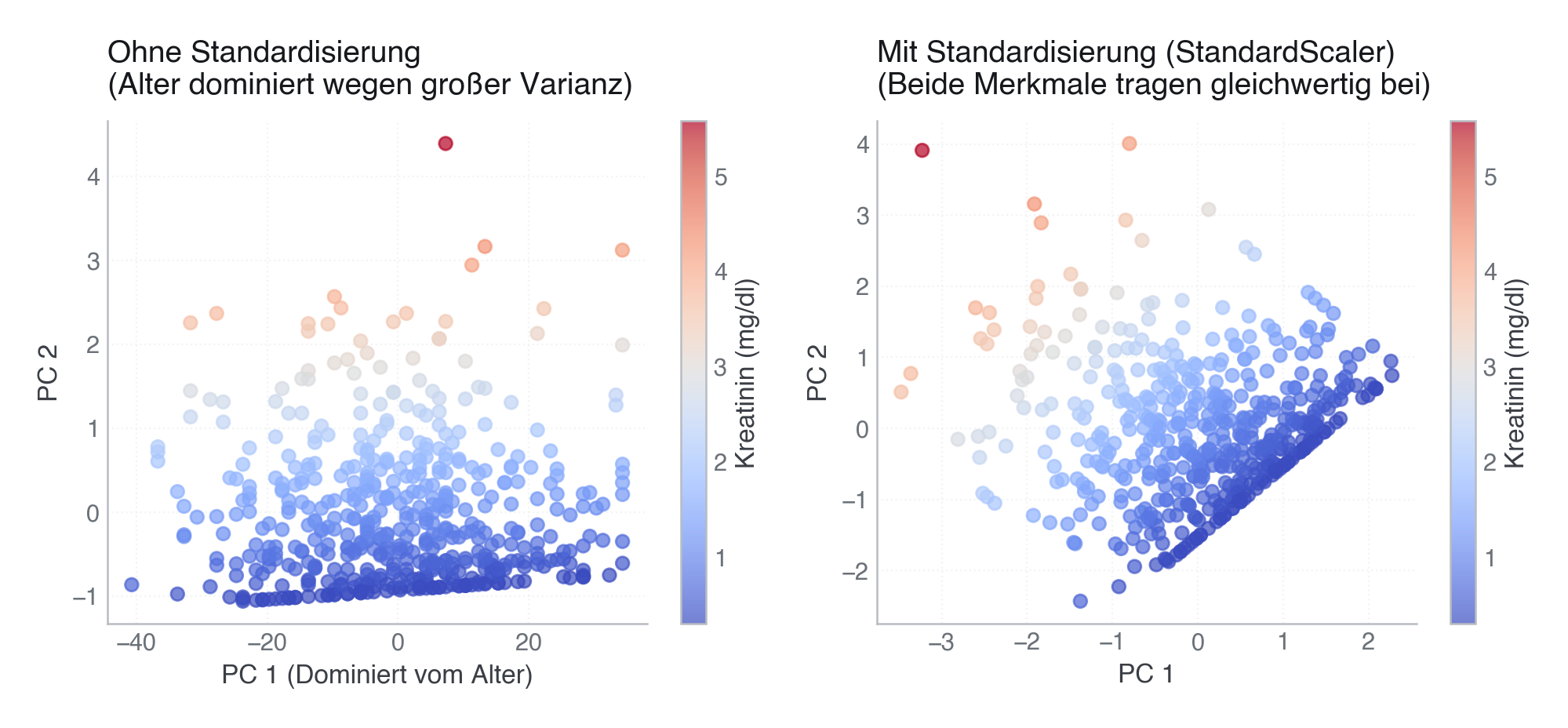
Wenn Merkmale unterschiedliche Einheiten und Skalen besitzen, dominiert die Variable mit der größten Varianz die Hauptkomponenten und das Clustering komplett.
- Ohne Standardisierung: Da das Alter (Spanne 20 bis 90 Jahre) viel größere Absolutwerte hat als Kreatinin (Spanne 0,5 bis 5,0 mg/dl), richtet sich die PCA (und damit auch K-Means) fast ausschließlich nach dem Alter aus. Das klinisch hochrelevante Kreatinin wird ignoriert (die Farben verschmieren zufällig quer über die y-Achse).
- Mit Standardisierung (StandardScaler): Beide Variablen tragen gleichwertig bei, und Kreatinin wird sauber entlang der Achsen getrennt.
In R:
library(tidyverse); library(recipes) labs <- read_csv("https://schradern.github.io/data-science-coach/data/labor.csv", show_col_types = FALSE) df <- read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv", show_col_types = FALSE) |> left_join(labs, by = "patient_id") numeric_cols <- c("alter","sofa_score","crp_mg_l","bmi", "leukozyten_g_l","kreatinin_mg_dl","laktat_mmol_l") rec <- recipe(~ ., data = df[numeric_cols]) |> step_impute_median(all_predictors()) |> step_normalize(all_predictors()) X_scaled <- rec |> prep() |> bake(new_data = NULL) |> as.matrix()
2 k-Means: k wählen mit Elbow und Silhouette
k-Means minimiert die Intra-Cluster-Varianz. Die Anzahl k lernt das Modell nicht, sie ist eine Wahl des Analysten.
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.from sklearn.cluster import KMeans from sklearn.metrics import silhouette_score inertias, silhouettes = [], [] K_RANGE = range(2, 9) for k in K_RANGE: km = KMeans(n_clusters=k, random_state=SEED, n_init=10) labels = km.fit_predict(X) inertias.append(km.inertia_) silhouettes.append(silhouette_score(X, labels)) # In dieser Kohorte ist der Silhouettenkoeffizient bei k=2 am höchsten (0,130), # knapp vor k=7 (0,123) und k=5 (0,122); k=3 liegt mit 0,115 sogar darunter. # Alle Werte liegen zwischen 0,10 und 0,13 -> schwache, überlappende # Clusterstruktur, kein k trennt die Kohorte sauber. Trotzdem wird unten # bewusst mit k=3 weitergearbeitet (drei Schweregrad-Stufen), eine klinisch # begründete, keine rein statistisch optimale Wahl (siehe Stolperstein). best_k = 3 km_final = KMeans(n_clusters=best_k, random_state=SEED, n_init=10).fit(X) df["cluster"] = km_final.labels_
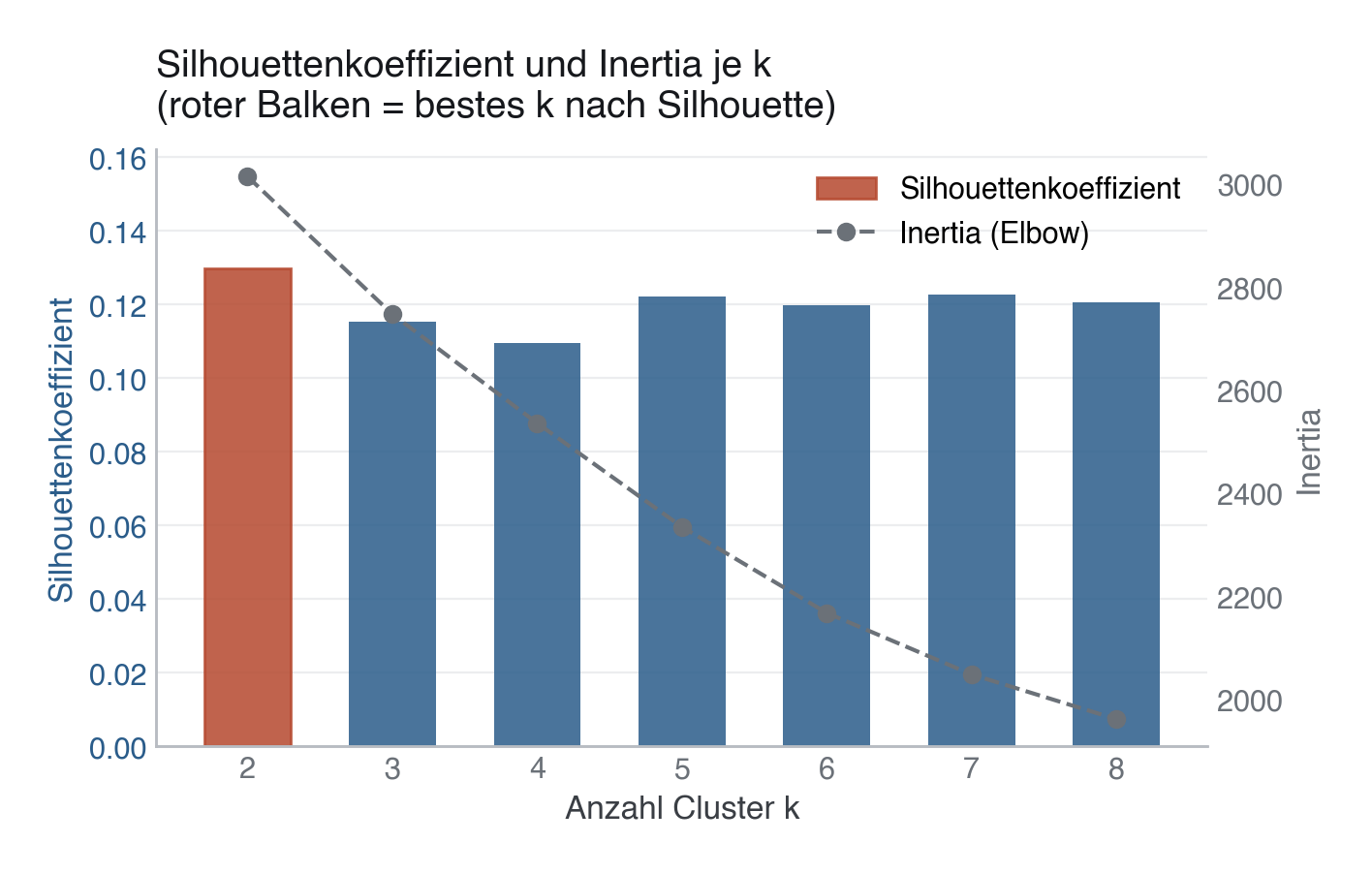
Der Silhouettenkoeffizient bevorzugt kompakte, gut getrennte Cluster, er belohnt nicht notwendigerweise klinisch sinnvolle Gruppen. Hier ist er sogar bei k=2 am höchsten (0,130) und nicht bei k=3 (0,115), das statistisch "beste" k muss trotzdem nicht das klinisch nützlichste sein: Zwei Cluster wären für eine differenzierte Risikostratifizierung oft zu grob. Wer k=3 wählt, muss das explizit klinisch begründen, nicht als "das Elbow/die Silhouette bestätigt 3" ausgeben, das wäre hier schlicht falsch.
Für FortgeschritteneVertiefung
k-Medoids (PAM) ist robuster gegenüber Ausreißern als k-Means und liefert echte Patientenrepräsentanten als Clusterzentren, hilfreich für die klinische Kommunikation.
k-Means setzt implizit voraus, dass Cluster im euklidischen Raum ungefähr kugelförmig, varianzgleich und ähnlich groß sind, es minimiert schließlich nur die Distanz zum jeweiligen Clusterzentrum. Längliche, unterschiedlich dichte oder stark unterschiedlich große Gruppen kann k-Means daher nicht sauber trennen, selbst nach Standardisierung. Passt die klinische Struktur nicht zu dieser Annahme, sind Gaussian Mixture Models (für elliptische, unterschiedlich orientierte Cluster), DBSCAN/HDBSCAN (für dichtebasierte Cluster beliebiger Form) oder hierarchisches Clustering mit Ward-Linkage (wenn die Verschmelzungsstruktur selbst interessiert) die geeigneteren Werkzeuge.
In R:
library(factoextra) fviz_nbclust(X_scaled, kmeans, method = "silhouette") km <- kmeans(X_scaled, centers = 3, nstart = 10, iter.max = 100) df$cluster <- km$cluster
3 Hierarchisches Clustering und Dendrogramm
Hierarchisches Clustering baut einen Baum aller Verschmelzungen. Du schneidest dort, wo ein großer Sprung im Abstand erscheint.
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.from scipy.cluster.hierarchy import linkage, dendrogram, fcluster import matplotlib.pyplot as plt Z = linkage(X, method="ward") # Cut at k=3 to match k-means labels_hier = fcluster(Z, t=3, criterion="maxclust")
Ward-Linkage minimiert die Varianz innerhalb von Clustern und setzt euklidische Distanz voraus, nach Standardisierung korrekt. Average- oder complete-Linkage reagieren anders auf Ausreißer und können sinnvoller sein, wenn die Daten nicht-sphärisch verteilt sind.
Für FortgeschritteneVertiefung
Ab etwa 500 Datenpunkten wird vollständiges hierarchisches Clustering rechenintensiv. BIRCH oder ein zweistufiger Ansatz (zuerst Mini-Batch-k-Means, dann Hierarchie auf Clusterzentren) skaliert besser.
In R:
dist_mat <- dist(X_scaled, method = "euclidean") hc <- hclust(dist_mat, method = "ward.D2") plot(hc, labels = FALSE, main = "Dendrogramm (Ward)") abline(h = 15, col = "red", lty = 2) # Schnittlinie bei k=3 df$cluster_hier <- cutree(hc, k = 3)
4 Dimensionsreduktion: PCA und UMAP
Hochdimensionale Daten lassen sich nicht direkt visualisieren. PCA und UMAP projizieren auf zwei Dimensionen, aber grundverschieden.
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.from sklearn.decomposition import PCA # PCA: linear, preserves global variance pca = PCA(n_components=2, random_state=SEED) X_pca = pca.fit_transform(X) explained = pca.explained_variance_ratio_ print(f"PC1+PC2 explain {explained.sum():.1%} of variance") # UMAP: non-linear, preserves local structure try: import umap reducer = umap.UMAP(n_components=2, random_state=SEED) X_2d = reducer.fit_transform(X) method_label = "UMAP" except (ImportError, TypeError) as exc: # ImportError: umap-learn not installed. TypeError: an installed # umap-learn can be incompatible with the installed scikit-learn version # (a real version-skew failure, not just a missing package). from sklearn.manifold import TSNE X_2d = TSNE(n_components=2, random_state=SEED, perplexity=30).fit_transform(X) method_label = "t-SNE (UMAP nicht verfügbar/inkompatibel)" print(f"Note: UMAP failed ({exc}), using {method_label}")
UMAP-Ergebnisse hängen von n_neighbors und min_dist ab. Zwei Läufe mit verschiedenen Seeds können verschiedene Layouts erzeugen, nie ohne Fixed Seed veröffentlichen und Hyperparameter immer berichten.
In UMAP ist nur die lokale Nachbarschaftsstruktur verlässlich. Der Abstand zwischen weit auseinanderliegenden Clustern, ihre relative Größe im Bild und ihre visuelle Dichte sind keine verlässlichen Maße für tatsächliche Ähnlichkeit, Gruppengröße oder Gruppendichte, UMAP verzerrt diese Größen algorithmisch beim Projizieren. Interpretiere nur, welche Punkte einander nahe liegen, nie wie weit Cluster auseinanderliegen oder wie groß/dicht sie erscheinen.
Für FortgeschritteneVertiefung
PCA erklärt, wie viel Varianz jede Komponente trägt (erklärte Varianz); UMAP erklärt das nicht. Für explorative Kommunikation ist UMAP visuell eindrücklicher, für reproduzierbare Erklärbarkeit ist PCA transparenter.
In R:
library(umap) # install.packages("umap") falls nicht vorhanden pca_res <- prcomp(X_scaled, scale. = FALSE) X_pca_r <- pca_res$x[, 1:2] # UMAP (Fallback: PCA nutzen, falls Paket fehlt) tryCatch({ umap_res <- umap(X_scaled) X_umap <- umap_res$layout }, error = function(e) { message("umap nicht verfügbar, nutze PCA-2D als Fallback") X_umap <<- X_pca_r })
5 Cluster als klinische Phänotypen profilieren
Cluster erhalten Bedeutung erst durch die Inhalte ihrer Mitglieder, nicht durch ihre Nummer.
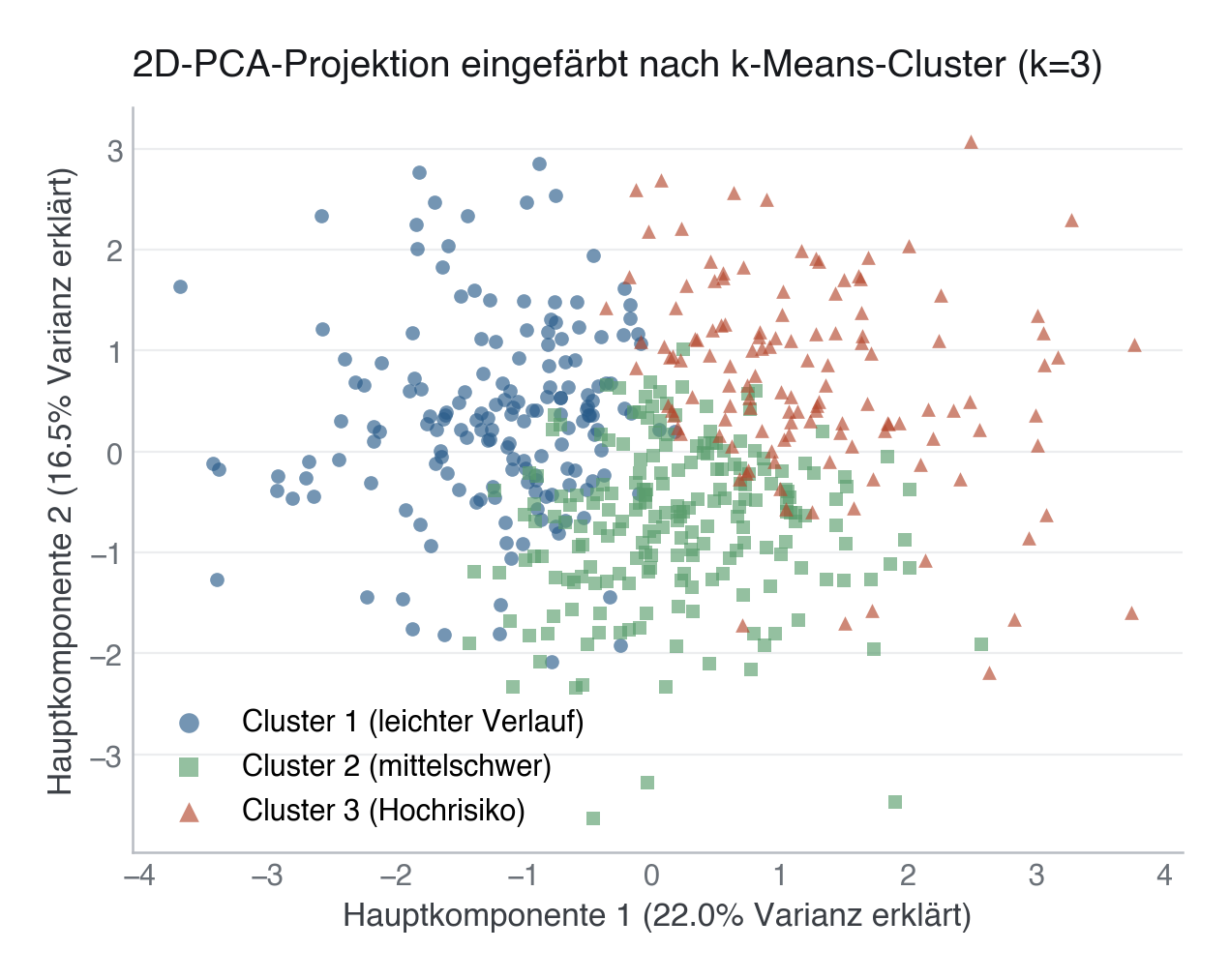
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.import pandas as pd profile = df.groupby("cluster")[NUMERIC + ["verweildauer_tage", "verstorben_30d"]].mean().round(2) print(profile.T) # Qualitative labels — assigned AFTER reading the profile, by ranking # clusters on mean SOFA (illness severity). k-Means cluster IDs (0, 1, 2, …) # are arbitrary and NOT guaranteed to come out in severity order (they don't, # for this cohort/seed: the highest-SOFA cluster is ID 1, not ID 2), so a # fixed {1: "mild", 2: "moderate", 3: "severe"}-style mapping keyed on raw # IDs would silently mislabel the phenotypes. severity_order = profile["sofa_score"].sort_values().index.tolist() phenotype_names = dict(zip(severity_order, ["Leichter Verlauf", "Mittelschwer", "Hochrisiko"])) df["phenotype"] = df["cluster"].map(phenotype_names)
Die Etikettierung ist interpretativ, nicht algorithmisch. Zwei Analysten können dieselben Cluster unterschiedlich benennen. Jede Benennung muss durch die Merkmalsmittelwerte belegt und im Paper explizit gerechtfertigt sein. Zusätzlich: Cluster-IDs sind arbiträr, sie nach fester Nummer statt nach den tatsächlichen Profilwerten zu benennen, ist ein leicht zu übersehener Fehler.
Für FortgeschritteneVertiefung
Validiere Phänotypen extern, etwa ob Cluster-Unterschiede in Verweildauer (verweildauer_tage) oder Mortalität mit unabhängigen klinischen Daten übereinstimmen. Interne Cluster-Güte (Silhouette) ist nicht dasselbe wie externe klinische Validität.
In R:
profile <- df |> group_by(cluster) |> summarise(across(c(alter, sofa_score, crp_mg_l, laktat_mmol_l, verweildauer_tage, verstorben_30d), mean, na.rm = TRUE)) print(profile)
Wann du Hilfe holst. Bevor du gefundene Cluster als reale klinische Phänotypen kommunizierst, brauchst du Stabilitäts- und Reproduzierbarkeitsprüfungen und idealerweise eine externe Kohorte — ob eine Subgruppe Biologie oder Artefakt ist, entscheidet nicht der Silhouettenkoeffizient allein, und diese Einordnung trifft man mit Methodik und Fachdisziplin gemeinsam.
Fallstricke und Merksätze
- Cluster sind keine Diagnosen. Sie sind datengetriebene Ähnlichkeitsgruppen, klinisch validieren, bevor du sie publizierst oder danach handelst.
- Skalierung ist Pflicht. k-Means auf rohen Werten wird von Merkmalen mit großer Varianz (z. B. CRP in mg/l) dominiert.
- k ist eine Wahl, kein Faktum. Elbow und Silhouette helfen, aber die finale Entscheidung ist klinisch zu begründen.
- UMAP-Layouts sind nicht reproduzierbar ohne Fixed Seed. Immer
random_statesetzen und Hyperparameter berichten. - Hierarchisches Clustering skaliert schlecht. Bei großen Kohorten (>5 000) besser Mini-Batch-k-Means oder BIRCH.