Teil 4 – Statistische Inferenz und medizinische Statistik
19 · Propensity Score Matching und Weighting
- Das Prinzip des Propensity Scores (Neigungsscore) zur Confounder-Kontrolle in Beobachtungsstudien erklären.
- Einen Propensity Score mittels logistischer Regression berechnen.
- 1:1 Nearest-Neighbor-Matching mit Caliper und Inverse Probability Weighting (IPW) vergleichen.
- Die Balance-Prüfung über die Standardized Mean Difference (SMD) interpretieren – und wissen, wann ein Matching nicht gut genug ist und nachgebessert werden muss.
- Erklären, warum man einen Mediator nicht adjustiert, wenn der Gesamteffekt einer Exposition interessiert.
Auf dieser Seite
- Klinischer Aufhänger
- 1 Was ist ein Propensity Score?
- 2 Propensity Score Matching (PSM) in Python
- 3 Propensity Score Matching in R
- 4 Balance-Check: Standardized Mean Difference (SMD)
- Was der Caliper in unseren Daten tatsächlich bringt
- 5 Der Effekt von Diabetes auf die 30-Tage-Mortalität: zwei Schätzer, zwei Estimands
- Fallstricke und Merksätze
- Selbstcheck
Klinischer Aufhänger
Im geteilten Kohortendatensatz interessiert uns die Frage: Wie stark erhöht Diabetes das 30-Tage-Mortalitätsrisiko insgesamt? Ärzt:innen können diese Frage nicht direkt aus einem rohen Gruppenvergleich beantworten, weil Diabetiker:innen in unserer Kohorte im Schnitt älter sind und häufiger männlich, hypertensiv oder aktive Raucher:innen – alles Faktoren, die das Sterberisiko unabhängig beeinflussen (Confounding). Mittels Propensity Score Matching erstellen wir eine synthetische Kontrollgruppe, in der Diabetiker:innen und Nicht-Diabetiker:innen bezüglich dieser Confounder nahezu identisch sind.
Nicht jede mit dem Outcome assoziierte Variable gehört in das Propensity-Score-Modell.
In dieser Kohorte gilt (siehe Modul 15, DAG): alter ist ein echter Confounder von Diabetes und
Sterblichkeit – adjustieren. sofa_score dagegen ist ein Mediator: Diabetes selbst erhöht den
SOFA-Score bei Aufnahme (schwererer Verlauf), und der SOFA-Score wirkt dann auf die Mortalität. Würden wir
nach sofa_score matchen oder adjustieren, würden wir einen Teil des tatsächlichen Diabetes-Effekts
wegrechnen und nur noch den direkten Effekt schätzen – nicht den Gesamteffekt, nach dem wir hier fragen.
1 Was ist ein Propensity Score?
Der Propensity Score (PS) ist die bedingte Wahrscheinlichkeit, dass ein:e Patient:in exponiert ist (hier: Diabetes hat), gegeben die beobachteten Confounder:
PS = P(Diabetes = 1 | Alter, Geschlecht, Hypertonie, Raucherstatus)
Zur Einordnung: In einer hypothetischen randomisierten Studie, in der die Exposition per Münzwurf zugeteilt würde, wäre der PS für alle Patient:innen 0,5. In Beobachtungsstudien schätzen wir den PS über eine logistische Regression, bei der die Exposition der binäre Outcome ist.
Stolperstein – ist Diabetes überhaupt eine „Behandlung"? Propensity-Score-Methoden imitieren ein randomisiertes Experiment. Das setzt eine wohldefinierte Intervention voraus: einen Eingriff, den man einer Person zuweisen könnte (Consistency-Annahme). Diabetes ist nicht manipulierbar – man kann niemandem „Diabetes" zuteilen. „Der kausale Effekt von Diabetes" ist ohne spezifizierte Intervention mehrdeutig: gemeint sein könnte eine Blutzuckersenkung, eine Diagnose, eine Krankheitsdauer – lauter verschiedene Kontraste (Hernáns Frage „does water kill?" – ertrinken oder verdursten?). Wir behalten das Beispiel hier als Illustration der Mechanik (PS, Matching, IPW, Balance) auf vertrauten Daten – die geschätzte Zahl ist aber kein sauber definierter Kausaleffekt. Ein verteidigbares Target Trial würde stattdessen eine konkrete Intervention emulieren, z. B. „intensive vs. Standard-Blutzuckerkontrolle ab Aufnahme", mit klaren Ein-/Ausschlusskriterien und einem definierten Zeitnullpunkt.
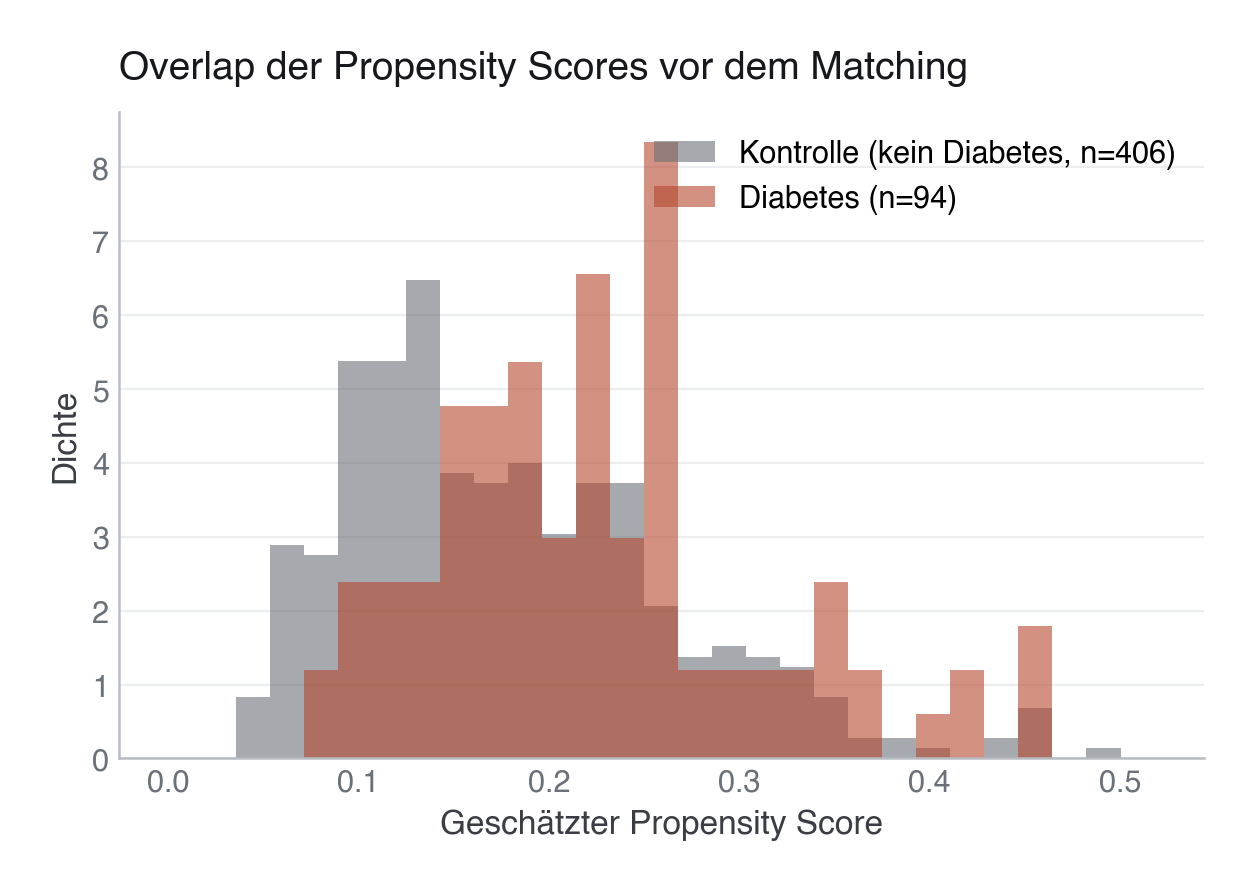
Stolperstein – Positivity. Kausale Kontraste brauchen zusätzlich, dass in jeder Confounder-Kombination
sowohl exponierte als auch nicht-exponierte Personen vorkommen können (0 < PS < 1). Fehlt in einer
Subgruppe eine der beiden Gruppen (Positivity-Verletzung), extrapoliert das Modell blind, und IPW erzeugt
extreme Gewichte. Der Overlap-/Common-Support-Check (unten) prüft genau das.
2 Propensity Score Matching (PSM) in Python
Wir berechnen den PS über eine logistische Regression und matchen 1:1 auf den Logit des PS – mit einem Caliper, der zu weit entfernte Matches ablehnt, statt sie zu erzwingen.
.py schreiben und mit dem ▶-Knopf in VS Code ausführen – oder Zeile für Zeile in die Python-Konsole. Setzt die in Modul 02 eingerichtete Umgebung voraus.import pandas as pd import numpy as np from sklearn.linear_model import LogisticRegression from scipy.spatial.distance import cdist # 1. Kohorte laden; Exposition = Diabetes, Confounder = Alter/Geschlecht/Hypertonie/Raucherstatus df = pd.read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv") df["geschlecht_m"] = (df["geschlecht"] == "maennlich").astype(int) df["raucher_aktiv"] = (df["raucherstatus"] == "aktiv").astype(int) covariates = ["alter", "geschlecht_m", "hypertonie", "raucher_aktiv"] # NICHT sofa_score (Mediator!) # 2. Propensity Score schätzen (unregularisierte logistische Regression) lr = LogisticRegression(max_iter=2000, penalty=None) lr.fit(df[covariates], df["diabetes"]) df["ps"] = lr.predict_proba(df[covariates])[:, 1] df["logit_ps"] = np.log(df["ps"] / (1 - df["ps"])) # 3. 1:1 Matching mit Caliper (0,2 x SD des Logit-PS - Faustregel nach Austin 2011) caliper = 0.2 * df["logit_ps"].std() treated = df[df["diabetes"] == 1] control = df[df["diabetes"] == 0] dists = cdist(treated[["logit_ps"]], control[["logit_ps"]]) matched_pairs, available = [], np.ones(len(control), dtype=bool) for i in np.argsort(dists.min(axis=1)): # beste Matches zuerst vergeben d = dists[i].copy(); d[~available] = np.inf j = int(np.argmin(d)) if d[j] <= caliper: matched_pairs.append((treated.index[i], control.index[j])) available[j] = False df_matched = pd.concat([df.loc[[t for t, _ in matched_pairs]], df.loc[[c for _, c in matched_pairs]]]) print(f"Gematchte Paare: {len(matched_pairs)} von {len(treated)} Diabetiker:innen")
🗣 Code-Verbalisierung:
LogisticRegression(penalty=None)trainiert eine unregularisierte logistische Regression (wie R'sglm()) für die Behandlungszuweisung (diabetes) basierend auf den vier Confoundern.cdistberechnet die absoluten Differenzen der Logit-Propensity-Scores zwischen allen Diabetiker:innen und allen Nicht-Diabetiker:innen.- Der Caliper (hier
0,2 × SD(logit PS), eine gängige Faustregel) verwirft Matches, die zu weit auseinanderliegen, statt schlechte Paare zu erzwingen – das kostet ein paar Patient:innen (Support-Verlust), verbessert aber die Balance. np.argsort(dists.min(axis=1))vergibt zuerst die eindeutigsten Matches, damit knappe Caliper-Fälle nicht durch Zufall den falschen Partner verlieren.
3 Propensity Score Matching in R
In R ist MatchIt der Standard für Matching-Analysen; die Caliper-Option ist eingebaut.
library(MatchIt) library(readr) df <- read_csv("https://schradern.github.io/data-science-coach/data/kohorte.csv") df$geschlecht_m <- as.integer(df$geschlecht == "maennlich") df$raucher_aktiv <- as.integer(df$raucherstatus == "aktiv") # Matching mit Caliper (0.2 x SD des Logit-PS), NICHT nach sofa_score (Mediator) m_out <- matchit(diabetes ~ alter + geschlecht_m + hypertonie + raucher_aktiv, data = df, method = "nearest", caliper = 0.2) summary(m_out) df_matched <- match.data(m_out)
4 Balance-Check: Standardized Mean Difference (SMD)
Nach dem Matching musst du prüfen, ob die Confounder zwischen den Gruppen ausgeglichen sind. Der p-Wert ist hierfür ungeeignet (da er von der Stichprobengröße abhängt). Stattdessen nutzen wir die Standardized Mean Difference (SMD):
SMD = (Mean_exponiert - Mean_kontrolle) / Pooled_SD
- |SMD| < 0,10: gilt als gute Balance – die Gruppen sind bezüglich dieses Merkmals weitgehend vergleichbar.
- |SMD| > 0,10: Hinweis auf verbleibendes Confounding. Das Matching sollte verfeinert werden (z. B. durch einen Caliper, der maximale Score-Unterschiede begrenzt).
Die Schwelle 0,10 ist eine Konvention, kein Testergebnis. Sie stammt aus der
Simulationsliteratur (Austin 2011, dieselbe Quelle wie der Caliper 0,2 × SD) und beschreibt eine als
praktisch akzeptabel geltende Restimbalance – sie ist kein statistischer Test und kein Beweis für
Unverzerrtheit. Eine SMD knapp unter 0,10 in einer kleinen Kohorte schließt relevantes
Restconfounding nicht aus. Berichte die SMD als stetige Größe, nicht als bestanden/durchgefallen.
Was der Caliper in unseren Daten tatsächlich bringt
code/python.py (und code/r.R) rechnen das komplett durch – erst ohne, dann mit Caliper, und
zusätzlich mit IPW als drittem Ansatz:
| Confounder | SMD vor Matching | SMD nach Caliper-Matching | SMD IPW-gewichtet |
|---|---|---|---|
| Alter | 0,58 | −0,03 ✓ | 0,13 ✗ |
| Geschlecht (männl.) | −0,15 | −0,06 ✓ | 0,03 ✓ |
| Hypertonie | 0,17 | 0,00 ✓ | 0,02 ✓ |
| Raucher:in (aktiv) | 0,08 | 0,03 ✓ | 0,03 ✓ |
Mit Caliper (93 von 94 Diabetiker:innen matchbar – eine Person hat keinen ausreichend ähnlichen Partner und fällt heraus) erreichen alle vier Confounder die 0,10-Schwelle. Ohne Caliper verfehlt „Geschlecht" die Schwelle knapp (SMD ≈ 0,11–0,15 je nach Zufallsreihenfolge) – genau der Fall, den die Fallstricke unten beschreiben. IPW balanciert Geschlecht, Hypertonie und Raucherstatus gut, lässt aber Alter bei SMD ≈ 0,13 – ein Hinweis, dass die Gewichte hier (Bereich 1,0–13,1) allein nicht ausreichen und Trimming oder ein stabilisiertes Gewicht sinnvoll wäre.
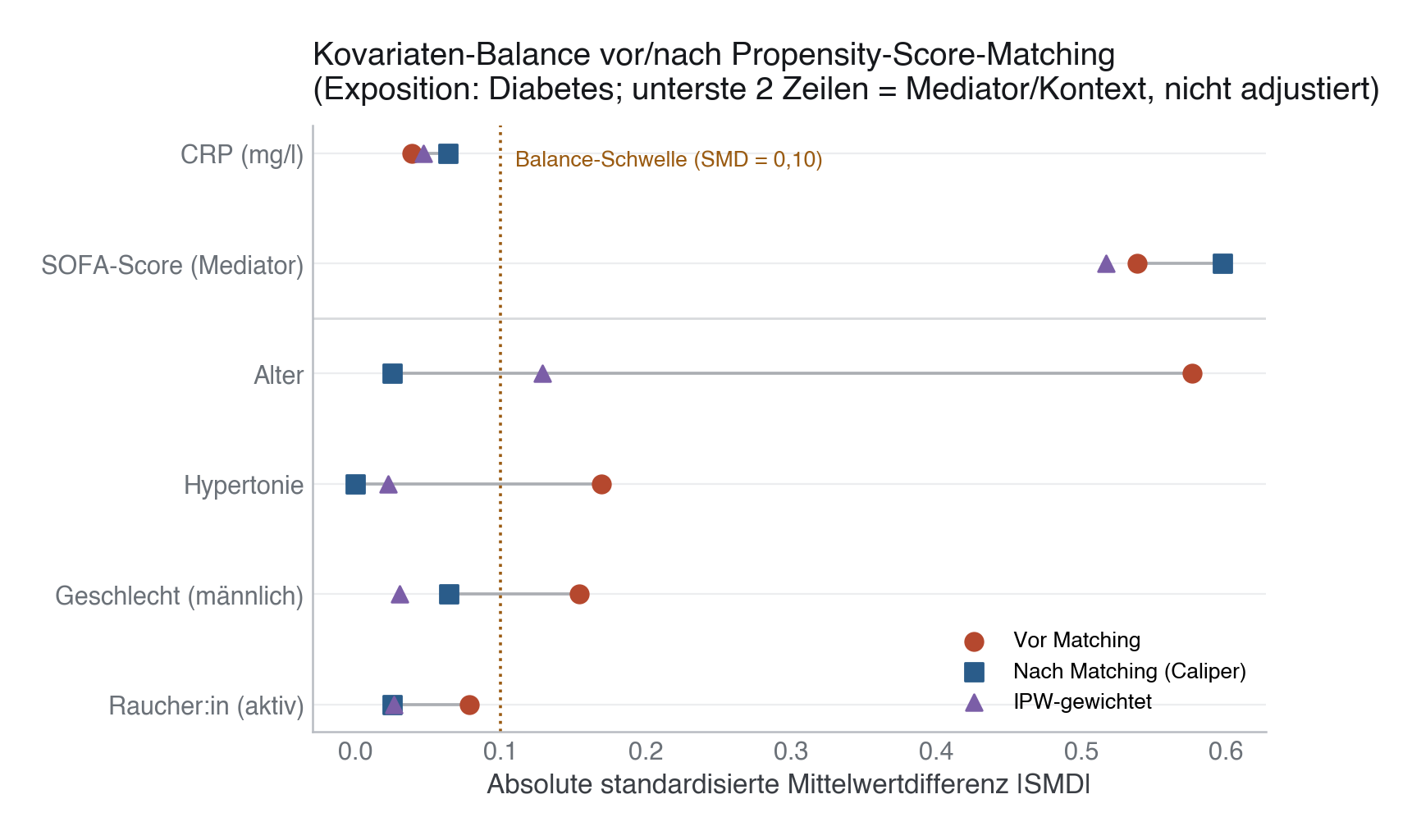
Der SOFA-Score bleibt in allen drei Ansätzen deutlich unbalanciert (SMD 0,52–0,60) – das ist kein Fehler, sondern folgt direkt aus der DAG-Logik: Diabetes verursacht ursächlich höhere SOFA-Werte bei Aufnahme. Balance auf einem Mediator zu erzwingen würde einen Teil des wahren Diabetes-Effekts eliminieren.
5 Der Effekt von Diabetes auf die 30-Tage-Mortalität: zwei Schätzer, zwei Estimands
| Methode | Estimand | Odds Ratio | 95 %-CI |
|---|---|---|---|
| Roh (unadjustiert) | – (nur Assoziation) | 2,07 | [1,19; 3,58] |
| Gematcht (Caliper), 93 Paare | ATT (Effekt bei den Behandelten) | 1,39 | [0,71; 2,70] |
| IPW-gewichtet | ATE (Effekt in der Gesamtkohorte) | 1,86 | [1,03; 3,36] |
Stolperstein – zwei verschiedene Zielgrößen, nicht nur zwei Genauigkeiten. Matching 1:1 auf die
Behandelten schätzt den ATT (Average Treatment effect on the Treated) – den Diabetes-Effekt in der
Population der 93 gematchten Diabetiker:innen. IPW mit 1/PS bzw. 1/(1−PS) gewichtet die gesamte
Kohorte auf und schätzt den ATE (Average Treatment Effect) – den Effekt, als hätte man die ganze
Kohorte exponiert bzw. nicht exponiert. ATT und ATE stimmen nur überein, wenn der Effekt in allen
Subgruppen gleich groß ist; unterscheiden sich Behandelte und Gesamtkohorte in effektmodifizierenden
Merkmalen, dürfen die Zahlen echt auseinanderlaufen – die Lücke zwischen 1,39 und 1,86 ist also nicht
allein „Zufall/Präzision".
Beide lassen den Mediator SOFA-Score bewusst aus und schätzen daher jeweils einen Gesamteffekt – nur auf unterschiedlichen Zielpopulationen. Der rohe Vergleich überschätzt das Risiko am stärksten; nach Confounder-Kontrolle sinkt die Punktschätzung.
Stolperstein – die richtige Varianz. Die Konfidenzintervalle oben stammen aus den korrekten Fehlerschätzern, nicht aus den Voreinstellungen:
- Gematcht (ATT): Die beiden Personen eines Matched Pairs sind nicht unabhängig. Der Standard-SE
eines gewöhnlichen Logit-Modells behandelt sie fälschlich als unabhängig. Korrekt ist ein
cluster-robuster SE auf der Paar-ID (
cov_type="cluster"in Python bzw.sandwich::vcovCL()in R). Das breite CI liegt hier tatsächlich an der kleinen Fallzahl (nur 2 × 93 Personen) – aber das entlässt einen nicht aus der Pflicht, die Paarung im SE zu berücksichtigen. - IPW (ATE): Nicht-ganzzahlige IPW-Gewichte sind keine Häufigkeiten. Behandelt man sie als
freq_weights(Python) oder mitfamily = quasibinomialund Modell-SE (R), bläht das die Pseudo-Fallzahl auf ≈ 2 × N auf bzw. schätzt einen Dispersionsparameter – beides ergibt ein zu schmales, antikonservatives CI ([1,35; 2,57]schließt die 1 dann fälschlich sicher aus). IPW braucht einen robusten (Sandwich-)Schätzer:var_weights+cov_type="HC0"in Python,lmtest::coeftest(fit, vcov = sandwich::vcovHC(fit, "HC0"))in R. Das breitere CI[1,03; 3,36]schließt die 1 nur noch knapp aus.
Darüber hinaus – Sandwich vs. Bootstrap. Der Sandwich-SE ignoriert, dass der Propensity Score selbst geschätzt wurde; für den ATE ist er dadurch tendenziell leicht konservativ. Der Goldstandard ist ein nichtparametrischer Bootstrap, der in jeder Wiederholung PS- und Outcome-Modell neu schätzt. Auf diesen Daten liegen beide praktisch gleichauf (Sandwich-SE des log-OR ≈ 0,302 vs. Bootstrap ≈ 0,305 über 2000 Resamples) – deshalb ist der Sandwich hier vertretbar. Bei starken Gewichten oder komplexeren PS-Modellen sollte man den Bootstrap bevorzugen.
Extreme Gewichte, Stabilisierung, Trimming. Die unstabilisierten Gewichte reichen von 1,04 bis 13,08 –
ein paar wenige Patient:innen dominieren die Schätzung. Stabilisierte Gewichte (P(A=a)/P(A=a|X))
schätzen denselben ATE, schrumpfen den Bereich aber auf 0,41–2,46; Punktschätzung und CI bleiben hier
unverändert (1,86 [1,03; 3,36]). Auch Trimming beim 99. Perzentil (Kappung bei 9,78) ändert kaum etwas
(1,83 [1,02; 3,29]) – ein beruhigendes Zeichen, dass das Ergebnis nicht von einzelnen Extremgewichten
getragen wird.
Wann du Hilfe holst. Sobald die Überlappung (Positivität) fraglich ist, unklar bleibt, ob du ATT oder ATE schätzen willst, oder plausibel ungemessene Confounder bestehen, hol methodische Beratung — kein Propensity-Score repariert ein schwaches Design allein.
Fallstricke und Merksätze
Ungemessene Confounder. Propensity Scores balancieren nur gemessene Kovariaten aus. Wenn ein wichtiger Faktor (z. B. Ernährungsstatus oder klinischer Gesamteindruck) nicht dokumentiert ist, bleibt die Assoziation verzerrt. Nur ein RCT schützt zuverlässig vor ungemessenem Confounding.
Support-Verlust (Overlap). Wenn exponierte und Kontroll-Patient:innen völlig unterschiedliche Scores haben (kein Overlap), können viele Patient:innen nicht gematcht werden – in unserem Beispiel verliert der Caliper eine von 94 Diabetiker:innen. Das reduziert die Stichprobengröße und schränkt die Generalisierbarkeit auf den Bereich mit „Common Support" ein.
Für FortgeschritteneVertiefung
Selbst mit Caliper balanciert 1:1-PS-Matching nicht automatisch jede einzelne
Kovariate – es matcht auf den (skalaren) Score, nicht auf jede Variable einzeln. Prüfe deshalb immer die
SMD pro Kovariate, nicht nur die des Gesamtscores. Bei einer hartnäckig unbalancierten Variable helfen
exaktes Matching auf genau diese Variable (z. B. exact = "geschlecht_m" in MatchIt) oder
Mahalanobis-Distanz-Matching als Ergänzung.
Berichte in Publikationen immer die SMD vor und nach dem Matching in einer standardisierten Tabelle (Love Plot, wie oben) – das ist mittlerweile Standard in Zeitschriften mit Beobachtungsstudien.